This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License. This means that you are able to copy, share and modify the work, as long as the result is distributed under the same license.
Network Visualization and Analysis with Cytoscape: Enrichment Map from g:Profiler results
By Veronique Voisin
Goal of the exercise:
Create an EnrichmentMap and navigate through the network
During this exercise, you will learn how to create an EnrichmentMap from gene-set enrichment results. The enrichment tool chosen for this exercise is g:Profiler but an enrichment map can be created from various gene-set tools using the generic format or the more specific GSEA or BinGO interface.
Data
The data used in this exercise is gene expression data obtained from high throughput RNA sequencing used in Module2 . They correspond to Ovarian serous cystadenocarcinoma samples. We use precomputed results of the g:Profiler analysis performed in module 2 to create an Enrichment Map with the aim to transform the tabular format in a network to better visualize the relationships between the significant gene-sets.
EnrichmentMap
-
A red circle (node) is a pathway specific of the mesenchymal type.
-
Green edges
represent genes in common between 2 pathways (nodes).
-
A cluster of nodes represent overlapping and related pathways and may represent a common biological process.
- Clicking on a node will display the genes included in each pathway.
Start the exercise
To start the lab practical section, first download the files.
Four files are needed to create the enrichment map for this exercise (please download these files on your computer):
Expression: MesenchymalvsImmunoreactive_RNAseq_expression.txt
Enrichments: gprofiler_results_mesenchymal.txt
Ranks: MesenchymalvsImmunoreactive_RNAseq_ranks.rnk
Classes: MesenchymalvsImmunoreactive_RNAseq_classes.cls
Step 1
Launch Cytoscape and open Enrichment Map App
1a. Double click on Cytoscape icon
1b. Open Enrichment Map App
-
In the top menu bar:
-
Click on Apps -> Enrichment Map -> Create Enrichment Map
Step 2
Create an EnrichmentMap
2a. Locate the Control Panel on the left side of the window, select the Enrichment Map input panel to load enrichment results.
2b. Specify the following parameters:
-
in Analysis Type select generic (ex:gProfiler)
-
in User Input : (load results files by clicking on the respective (…) button and selecting the file)
- GMT - not required. This field should remain empty. TIP: If you are creating an Enrichment Map with generic results from some other program and it lacks a column listing the genes belonging to each set then you need to specify a gmt file outlining the genes belonging to each set.
- Expression - MesenchymalvsImmunoreactive_RNSseq_expression.txt TIP: This field is optional but recommended
-
Enrichments - gprofiler_results_mesenchymal.txt TIP: only field that is not optional
-
In Advanced box:
-
Ranks - MesenchymalvsImmunoreactive_RNAseq_ranks.rnk TIP: This field is optional but recommended
-
Classes - MesenchymalvsImmunoreactive_RNAseq_classes.cls TIP: This field is optional but recommended
-
Phenotype - In the text boxes replace UP with Mesenchymal. Mesenchymal will be associated with red nodes because it corresponds to the positive phenotype in the gprofiler_results_mesenonly_ordered.
-
In the Parameters box:
-
Set Q-value cut off to 0.0001 TIP: to set threshold to very small numbers select Scientific Notation and set Q-value cut off to 1E-04
- Select Jaccard similarity cutoff and set the value to 0.25
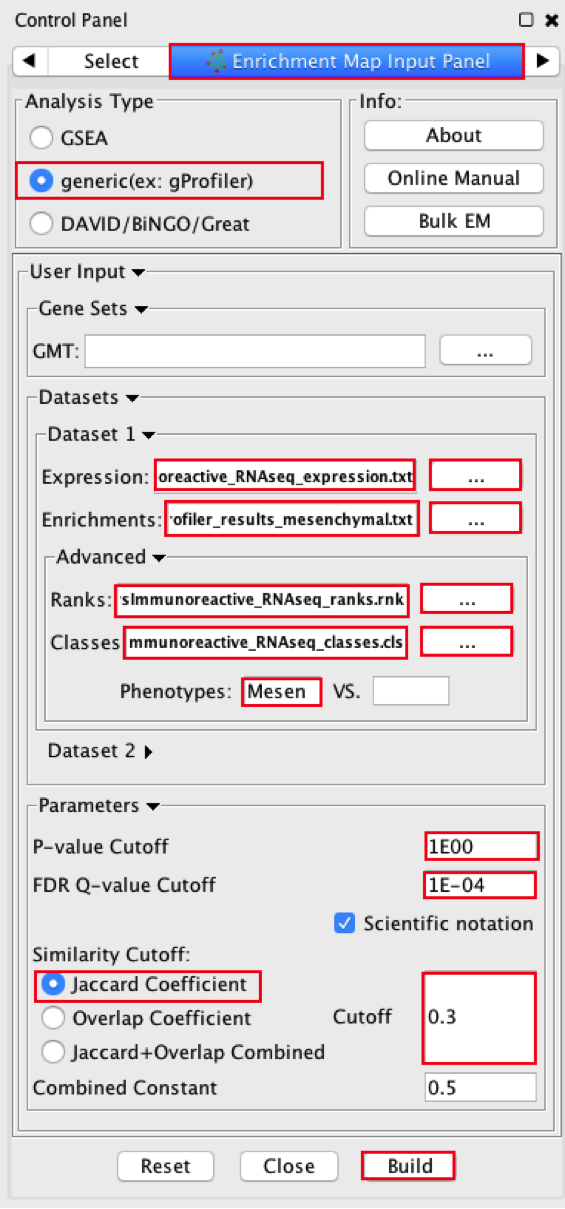
2c. Click on Build button at the bottom of the Enrichment Map Input panel.
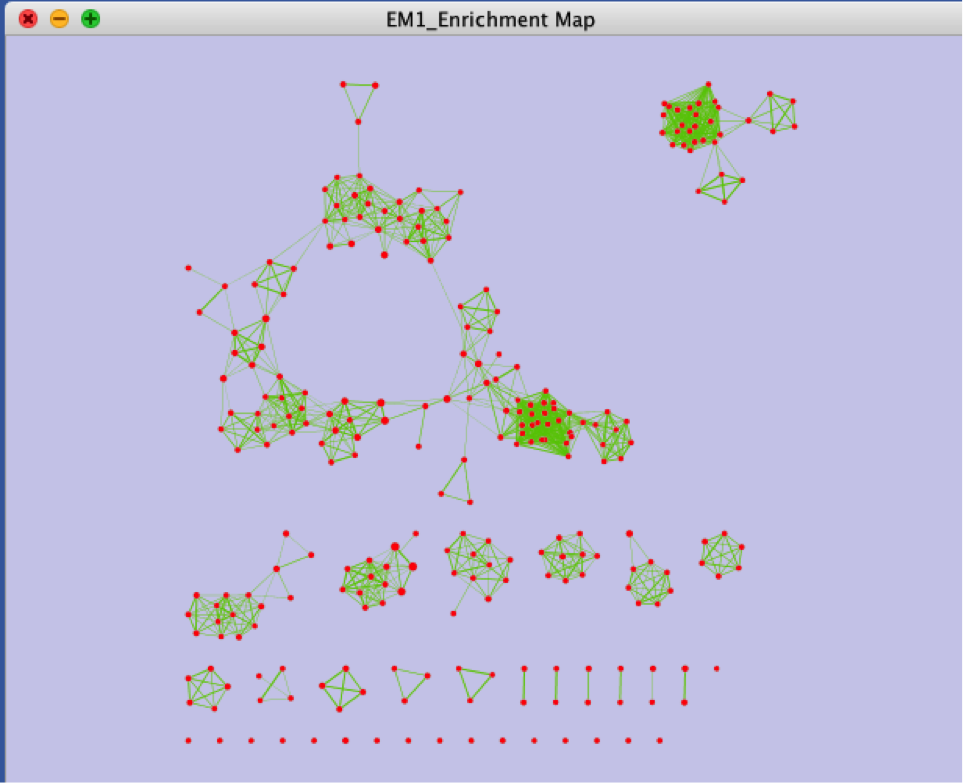
Unformatted results
Step 3
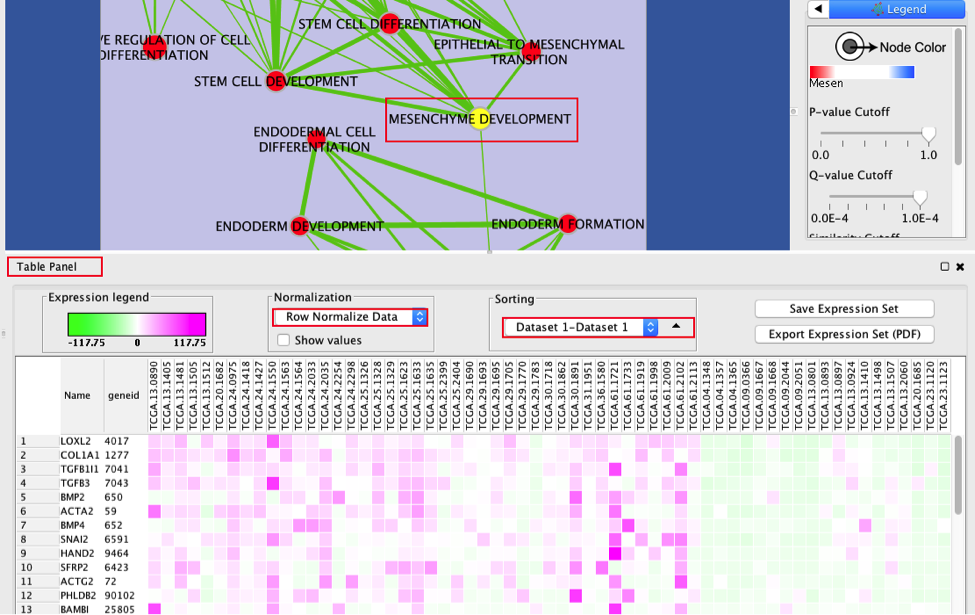
Select a node and visualize the Table Panel
3a. Click on a node
For this example the node MESENCHYME DEVELOPMENT has been selected.
When the node is selected, it is highlighted in yellow.
3b. In Table Panel:
-
Set Sorting to Dataset 1 - Dataset1
-
Set Normalization to Row Normalize Data
You can now visualize the genes in the MESENCHYME DEVELOPMENT pathway that are higher expressed in the mesenchymal samples when compared to the immunoreactive samples.