This work is licensed under a Creative Commons Attribution-ShareAlike 3.0 Unported License. This means that you are able to copy, share and modify the work, as long as the result is distributed under the same license.
Module 1.3 Tutorial: Metagenomic Data Resources
Prepared by William Hsiao (based on Rob Beiko’s material)
Introduction
While metagenomics researchers are mainly interested in studying the microbiome of a particular environment/host and generating data from new samples, it is still useful to know where to get existing datasets for comparative analysis or as a reference set (e.g. the human microbiome reference data). There is a huge amount of microbiome data available publically. Analyzing data and comparing results across multiple microbiome studies is still hindered by the variations in experimental protocols, sequencing processing pipelines, and inconsistencies in the metadata reported. Projects such as the EMP (http://www.earthmicrobiome.org/), OSD (https://www.microb3.eu/osd) and HMP (http://hmpdacc.org/) attempt to consolidate the experimental protocols and analysis pipelines used within these projects with limited success. Regarding metadata standardization, efforts such as MIMARKS checklist (minimum information about a marker gene sequence) and MIMS checklist (minimum information about a metagenome sequence) help establish the basic metadata that should be reported with each study. While the metadata fields have been established by these checklists, the data values are often inconsistent, for example, it’s not uncommon to see the same term spelled differently. Ontology (controlled vocabulary with clear definition and hierarchical relationships) efforts such as the OBO Foundry suite of ontologies provide some controlled vocabularies for metadata reporting.
This self-guided tutorial will serve as an introduction to various data repositories and the corresponding files they contain.
Note that in many cases these public repositories cannot take the strain of many simultaneous connections, and can even refuse connections if too much load is originating from one source.
In general, you should not worry about querying databases unless you are writing software for automated access these resources automatically.
1) NCBI SRA
NCBI Sequence Read Archive (SRA) is the main primary (raw data) repository for next generation sequence data (and synchronized with Europe and Japan repositories as well) and it has an overwhelming amount of raw sequence data. Included in this are marker-gene and metagenomic survey data from a remarkable range of projects. Understanding how the data are organized within SRA can help you navigate. The organization of SRA is as follows:
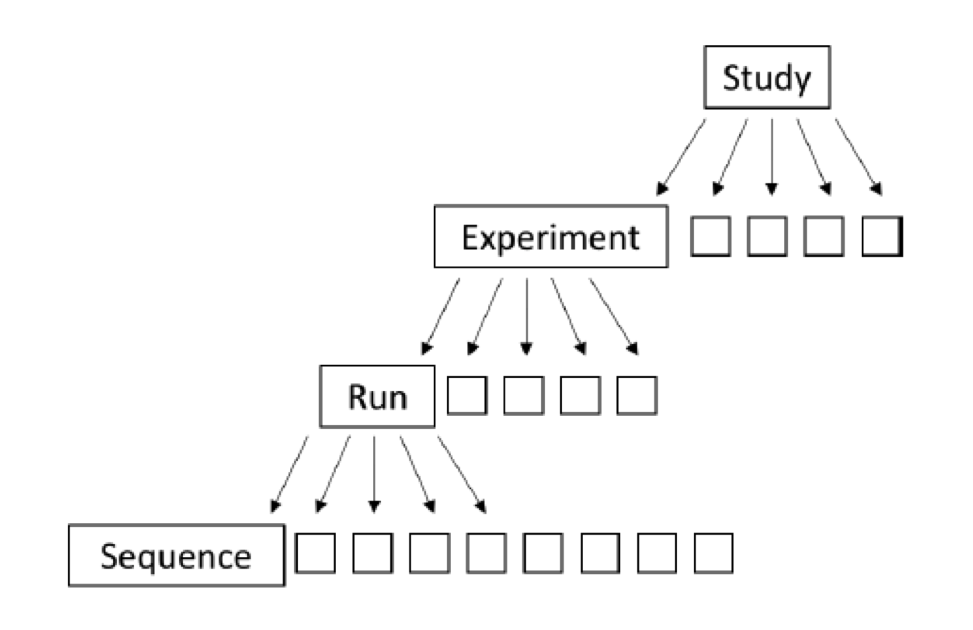
Study – A study is a set of experiments and has an overall goal. Studies are organized into Bioprojects.
Experiment – An experiment is a consistent set of laboratory operations on input material with an expected result.
Sample – An experiment targets one or more samples. Results are expressed in terms of individual samples or bundles of samples as defined by the experiment.
Run – Results are called runs. Runs comprise the data gathered for a sample or sample bundle and refer to a defining experiment. Each Run produces numerous sequences.
You can browse the data (and metadata) via a web browser but to download the actual sequence data, you will need to use SRA-toolkit, a command line tool.
Navigate to the SRA homepage (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=announcement). Click on “Browse” to look at the different studies available. There are over 71K records. While you can type in keywords in the search field, the datasets are not organized, making comprehensive search for particular data types (say 16S amplicon) difficult. A more common way to use SRA is to first identify the paper which contains the datasets that you are interested in. In the paper, a SRA ID and Bioproject ID will be provided to help you retrieve the raw data. We will focus on a particular study by typing 16S robustness into the search field (note that the keyword 16S is not an exhaustive nor specific way to identify all 16S marker gene microbiome studies in SRA).
Each study in this set of 18 (refer to the paper here: http://dnaresearch.oxfordjournals.org/content/20/3/241.long) comes from a different individual in a probiotic intervention study. The study page contains several cross-referencing links to various other databases (such as Bioproject) at NCBI, but let’s stay within the SRA by clicking on “Runs”.
On the resulting page you will see a table that summarizes the nine runs for this individual. The first was a pre-probiotic (!?) administration, the next four are identified as during the administration (eight weeks, with samples collected every two weeks), and the final four are unlabeled but likely correspond to the post-probiotic phase as identified in the paper. With the SRA we can drill as deeply as we want to into these data.
Click on the Run “DRR003411” to bring up a summary of this specific sequencing run, which corresponds to one time-point from this single individual. We can go into the “Reads” tab to look at each individual read, including the quality scores and intensity graph. You can flip through the reads on this page. So far, we’ve shown you how to browse or search for specific piece of information on SRA, but to download the sequences associated to a run/sample/experiment/study/project, you need to use SRA Toolkit.
Download the correct version (Windows or Mac) of the tool to your laptop. If you run “Fastq-dump.exe DRR003411” from the Windows command prompt (or the corresponding program in Mac or Linux) you will get a FASTQ file from one run, corresponding to the reads, quality scores, etc. you saw on the web page. “More DRR003411.fastq” should give you the file contents.
More information about the toolkit can be found at http://www.ncbi.nlm.nih.gov/Traces/sra/?view=toolkit_doc.
2) MG-RAST
Reference: Meyer et al. (2008)
This is a popular web portal for depositing and analyzing your marker gene or metagenomics data. The analysis tools on this site is fairly outdated and known to report erroneous results. Our advice is not to use MG-RAST for analysis unless your samples are from well-characterized environments. Still, there are better tools that we will show you in this workshop. MG-RAST however contains many samples (>35K metagenomes) and you can download them via a web browser (very slow to navigate) or via a programming API.
To start, click on “Browse Metagenomes” on the main page. This loads a table consisting of the public available datasets from MG-RAST. You can use the drop down menus in the column headers to narrow down the list of projects. Look at the available selections in the biome column, you’ll notice that there are a few redundant terms for “aquatic biome” (aquatic biome, Aquatic biome, and Aquatic habitat). This is an example where ontology could help to standardize the metadata. Browse around the datasets on your own to see if there are any from similar environments as your studies.
By clicking on the name of a particular project, you’ll get a description of the project (some projects have much more information than others). Then by clicking on the sample name, you can get a summary of default analyses done on these samples by MG-RAST. As noted before, these default analysis results could be erroneous so take the results with a grain of salt. Because of that, we will not spend time looking at MG-RAST analysis results. To download data from MG-RAST, you can click on the “Download” icon associated with each sample. Several data types (both raw and processes) are available for download.
A more high-throughput approach to download data from MG-RAST is to use the programming API (Reference). Browsing long lists of metagenomes can be an enormous pain, and MG-RAST has an application programming interface (API) documented at http://api.metagenomics.anl.gov/api.html; this allows you to generate custom queries directly via an http request. There is a tremendous amount of information that can be retrieved through this API. Note too as you hover over different links on the analysis page that everything is retrieved through the API as well.
The default return type is a JSON structure which contains relevant metadata as well as the answer to your query. There are libraries for importing JSON structs into (for example) Python; you might also want to install an add-on such as JSONView for Firefox (https://addons.mozilla.org/en-us/firefox/addon/jsonview/) or Chrome to get slightly more readable results in your browser. Tabular results are returned as a BIOM-formatted file.
A separate tutorial shows you on to download data using MG-RAST API: http://adina-howe.readthedocs.io/en/latest/mgrast/.
3) HMP DACC
The human microbiome project data (16S, metagenome) and associated reference genome data sets can be downloaded from HMP DACC. There are two primary cohort types:
-
Center “Healthy Cohort”: This is a single cohort of 300 healthy individuals, each sampled at 5 major body sites (oral, airways, skin, gut, vagina) and up to three time-points. Each body site consisted of a number of body subsites, for a total of 15 to 18 samples per individual per time-point. See Microbiome Analysis for more information.
-
Demonstration Project “disease cohorts”: These 15 projects each have one or more cohorts aimed at studying specific health conditions. Each project developed sampling, processing, and 16S or whole metagenome shotgun sequencing approaches according to their condition of interest. These cohorts include both controls and affected individuals. See Impacts on Health for a brief description of each demonstration project.
There are three primary data types:
-
Reference microbial genomes: Most of these are not derived from specific cohorts
-
Whole metagenome shotgun (mWGS) sequence
-
16S metagenomic sequence
Raw sequence data could be download via NCBI SRA using the Toolkits. Reference genomes and Value-added (clean-up or annotated) data can be downloaded following web links.
4) OTHER RESOURCES
QIITA (Github repo is a new platform (you could setup your own repository) and repository for metagenomic data, built on QIIME, that hosts a large number (about 45,000) of studies from the Earth Microbiome Project. QIITA is fairly new and requires registration. The basic workflow involves selecting studies and submitting them for analysis (e.g., beta diversity).